
Navitoclax联合芦可替尼,为芦可替尼单药进展或反应不佳MF患者提供新选择
骨髓纤维化(MF)病程多样,以贫血、骨髓的纤维化、进行性脾肿大、髓外造血和向白血病发展为特征。患者中位总生存期(OS)差异较大,可以<2年也可以>10年。JAK1/2抑制剂芦可替尼和JAK2抑制剂Fedratinib已被FDA批准用于治疗不适合造血干细胞移植的中/高危MF患者。两项I期研究已证实Fedratinib可降低骨髓的纤维化程度和JAK2V617F等位基因突变负荷,而芦可替尼单药治疗对骨髓的纤维化程度的影响较小,在以往II期和III期临床试验中尚未明晰其对驱动突变的等位基因负荷的影响。此外有研究表明同时伴有≥3个突变或高分子风险(HMR)突变的患者,使用JAK/STAT抑制剂的结局较差。HMR突变包括ASXL1、EZH2、SRSF2、IDH1或U2AF1突变。综上,JAK抑制剂不能完全满足MF患者的治疗需求。
Navitoclax(ABT-263)是一种口服生物的小分子BCL-2抑制剂,是BCL-2的BH3结构域模拟物,具有高亲和力,Ki值小于1nmol/L。该药物可以与包括BCL-2、BCL-XL、BCL-W在内的多种BCL-2蛋白家族的抗凋亡蛋白结合,破坏它们与BIM等促凋亡因子的相互作用,从而促进恶性细胞的凋亡。有临床前数据表明芦可替尼和navitoclax具有协同抗肿瘤作用:①芦可替尼可以抑制BCL-XL和MCL-1表达,使navitoclax以较低剂量有效抑制其余BCL-XL和BCL-2活性,以促进细胞凋亡;②navitoclax可以使骨髓细胞对JAK2抑制剂重新敏感。基于此,有研究者开展了II期REFINE试验(NCT03222609),评估了navitoclax联合芦可替尼在芦可替尼单药治疗有进展或反应不佳的MF患者中的疗效性与安全性。
▉ 研究方法
REFINE试验是一项全球多中心、开放标签II期临床研究,根据JAK抑制剂使用情况,患者分为四个研究队列,该报告介绍了1a队列的研究结果。患者入组条件:≥18岁;符合动态国际预后评分系统(DIPSS)定义的中/高危MF,东部肿瘤协作组的体能状态评分(ECOG PS)为0-2;不愿意或不适合接受异基因造血干细胞移植;可触及的脾肿大(肋缘下5cm)或脾体积达到450cm3;在第一剂navitoclax使用的8周前,每日两次接受过≥10mg剂量稳定的芦可替尼,总计≥12周;血小板计数为≥100×109/L。排除标准:6个月内接受过脾照射;外周血或骨髓原始细胞>10%;接受过抗凝/抗血小板的药物(不包括每日一次≤100mg的阿司匹林和低分子肝素);既往接受过BCL-2的BH3结构域模拟物的药物。
纳入的患者继续使用稳定剂量的芦可替尼,如果血小板≥75×109/L,则以每日一次50mg的起始剂量开始口服navitoclax,每周提高一次剂量直至最高剂量(300mg)。如果出现任何≥2级的出血事件,中断或停止navitoclax使用。出现4级中性粒细胞减少(中性粒细胞绝对值<0.5×109/L)、发热性中性粒细胞减少或3级非血液学毒性事件时,中断navitoclax和芦可替尼的使用。主要疗效终点为第24周时脾脏体积较基线水平减小≥35%(SVR35)的患者比例。次要疗效终点为第24周时,总体症状评分较基线水平降低≥50%(TSS50)的患者比例、血红蛋白的改善程度、骨髓的纤维化程度改善程度、安全性特征。
▉ 研究结果
患者基线特征
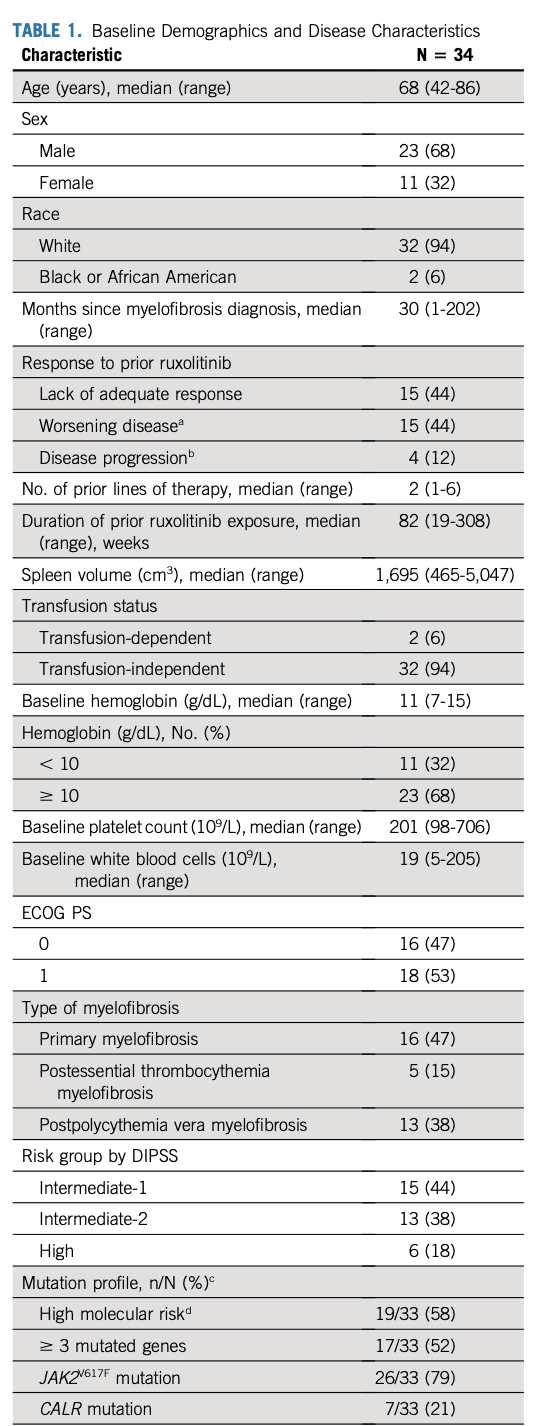
数据截止日期为2020年8月30日。34例MF患者接受了≥1剂navitoclax+芦可替尼。大部分患者为男性(n=23,68%),中位年龄68(范围:42-86)岁。先前接受芦可替尼治疗的中位持续时间为82(范围:19-308)周。大多数患者对先前芦可替尼的治疗反应不佳或出现疾病恶化,两种情况各为15例(44%);其余4例(12%)出现疾病进展(脾脏进展)。33例患者入组时进行了突变基因检查,26例(79%)有JAK2V617F突变,其余7例(21%)有CALR突变(4个1型和3个2型突变),无cMPL或TP53突变。17例患者(52%)有3个突变基因,19例(58%)有HMR突变;两例U2AF1突变的患者中有一例发生Q157点突变。具体特征如表1。

有效性分析
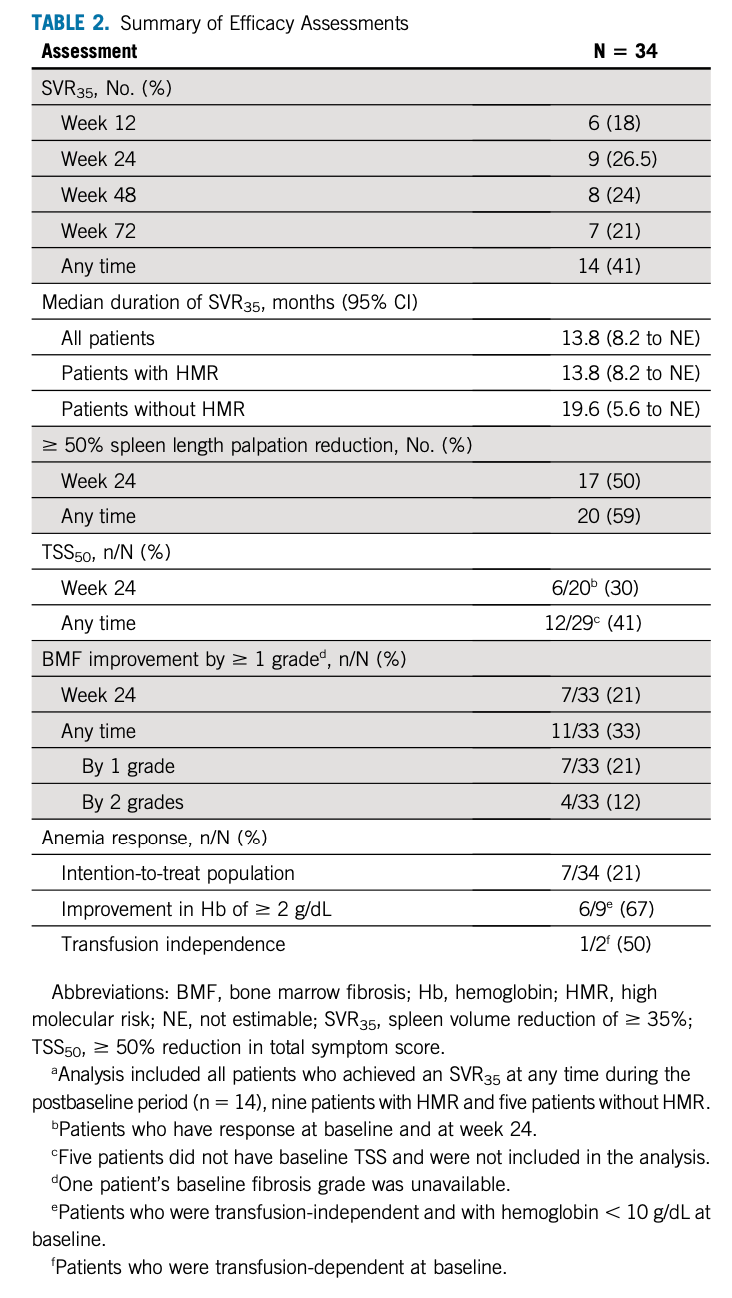
9例(26.5%)患者在第24周时达到SVR35。共有14例(41%)患者在研究期间的任意时间(不包括第24周)达到SVR35,其中6例(18%)和8例(24%)患者分别在第12周和第48周达到SVR35。有7例(22%)直至第72周才观察到SVR35。SVR35估计的中位持续时间为13.8个月(95%CI,8.2-无法预估[NE]),有HMR突变(n=9)和无HMR突变(n=5)组的SVR35无统计学差异。在第24周时,17例(50%)患者脾脏长度较基线水平明显减少50%,20例(59%)患者在研究期间的任意时间达到此状态。中位随访时间为21.6(范围:6.7-28.9)个月。中位OS未达到(NR)(95%CI,26.1-NE),24个月时的估计OS率为84%(95%CI,63.0-93.9)。有HMR突变(n=19)和无HMR突变(n=14)组的OS无显著差异。在第24周时,20例TSS可评估的患者中,6例(30%)达到TSS50,29例TSS可评估的患者中,12例(41%)在研究期间的任意时间达到TSS50。33例有骨髓纤维化程度基线结果的患者中,7例(21%)在24周时达到≥1级缓解,有11例(33%)在研究期间的任意时间达到≥1级缓解,其中7例为1级缓解,4例为2级缓解。11例(33%)患者的基线血红蛋白<10g/dL,其中7例(21%)血红蛋白改善了≥2g/dL;2例患者输血依赖,其中1例在治疗后脱离了输血依赖。具体疗效评估如表2。
安全性分析
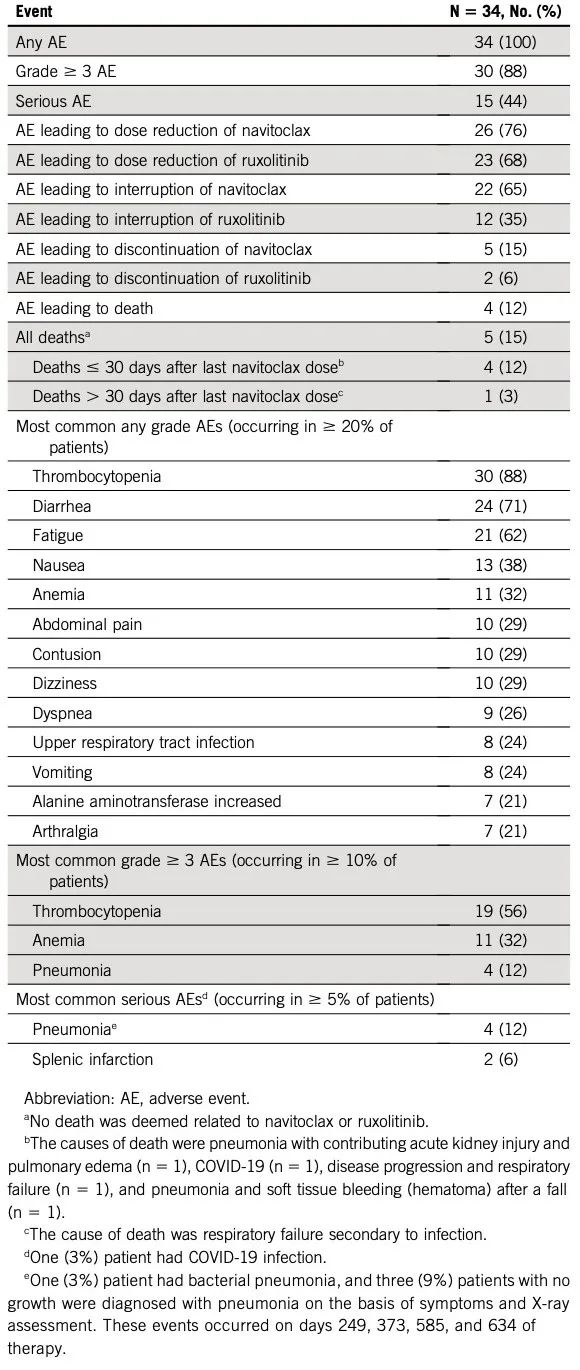
Navitoclax和芦可替尼两种药物的中位暴露时间分别为81周(范围:4-126)和83周(范围:10-124)。24例(71%)患者达到navitoclax最大使用剂量(300mg),每日一次;8例(24%)患者达到200mg,每日一次;1例(3%)达到100mg,每日一次;1例(3%)达到50mg,每日一次。26例(76%)患者因不良事件(AE)减低了navitoclax剂量,22例(65%)中断用药;减药和停药最常见原因是血小板减少,分别占19例(56%)。常见的任何级别AE包括血小板减少(n=30,88%)、腹泻(n=24,71%)、疲劳(n=21,62%)和恶心(n=13,38%);常见的3级AE是血小板减少(无临床显著出血;n=19,56%)、贫血(n=11,32%)和肺炎(n=4,12%);常见的严重AE是肺炎(n=4,12%)和脾梗死(n=2,6%)。具体如表3。
▉ 研究结论
该II期研究结果表明,navitoclax联合芦可替尼在对单药芦可替尼反应不佳或疾病恶化的患者群体中疗效是持久可控的。目前,研究者进一步开展了III期临床研究,并通过等位基因负荷和生物标志物修饰分析,来充分评估这两种新机制药物联合治疗的潜力。
参考文献:
Claire N. Harrison, Jacqueline S. Garcia, Tim C. P. Somervaille, et al. Addition of Navitoclax to Ongoing Ruxolitinib Therapy for Patients With Myelofibrosis With Progression or Suboptimal Response: Phase II Safety and Efficacy. Journal of Clinical Oncology. February 18, 2022.

 微信扫一扫
微信扫一扫 